Log in or Sign up
to access My Account functionalities
![]() Find similiar products:
Find similiar products:
-
By ATC code

![]() Find similiar products:
Find similiar products:
![]() This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 for how to report adverse reactions.
This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 for how to report adverse reactions.
ASPAVELI 1 080 mg solution for infusion
Each 20 mL vial contains 1 080 mg of pegcetacoplan.
Each mL contains 54 mg of pegcetacoplan.
Excipients with known effect
Each mL contains 41 mg of sorbitol.
Each vial contains 820 mg of sorbitol.
For the full list of excipients, see section 6.1.
Solution for infusion.
Clear, colourless to slightly yellowish aqueous solution with pH 5.0.
ASPAVELI is indicated in the treatment of adult patients with paroxysmal nocturnal haemoglobinuria (PNH) who are anaemic after treatment with a C5 inhibitor for at least 3 months.
Therapy should be initiated under the supervision of a healthcare professional experienced in the management of patients with haematological disorders. Self-administration and home infusion should be considered for patients who have tolerated treatment well in experienced treatment centres. The decision of a possibility of self-administration and home infusions should be made after evaluation and recommendation from the treating physician.
Posology
Pegcetacoplan can be given by a healthcare professional, or administered by the patient or caregiver following proper instruction.
Pegcetacoplan is administered twice weekly as a 1 080 mg subcutaneous infusion with a commercially available syringe system infusion pump that can deliver doses up to 20 mL. The twice weekly dose should be administered on Day 1 and Day 4 of each treatment week.
PNH is a chronic disease and treatment with ASPAVELI is recommended to continue for the patient's lifetime, unless the discontinuation of this medicinal product is clinically indicated (see section 4.4).
Patients switching to ASPAVELI from a C5 inhibitor
For the first 4 weeks, pegcetacoplan is administered as twice weekly subcutaneous doses of 1 080 mg in addition to the patient's current dose of C5 inhibitor treatment to minimise the risk of haemolysis with abrupt treatment discontinuation. After 4 weeks, the patient should discontinue C5 inhibitor before continuing on monotherapy with ASPAVELI.
Dose adjustment for ASPAVELI
The dosing regimen may be changed to 1 080 mg every third day (e.g., Day 1, Day 4, Day 7, Day 10, Day 13, and so forth) if a subject has a lactate dehydrogenase (LDH) level greater than 2 x upper limit of normal. In the event of a dose increase, LDH should be monitored twice weekly for at least 4 weeks (see section 4.4).
Missed dose of ASPAVELI
If a dose of pegcetacoplan is missed, it should be administered as soon as possible, then the regular schedule should be resumed.
Special populations
Elderly (>65 years old)
Although there were no apparent age-related differences observed in clinical studies, the number of patients aged 65 and over is not sufficient to determine whether they respond differently from younger patients. There is no evidence indicating any special precautions are required for treating an elderly population.
Renal impairment
Severe renal impairment (creatinine clearance <30 mL/min) had no effect on the pharmacokinetics (PK) of pegcetacoplan; therefore, pegcetacoplan dose adjustment in patients with renal impairment is not necessary. There are no data available for the use of pegcetacoplan in patients with end-stage renal disease (ESRD) requiring haemodialysis (see section 5.2).
Hepatic impairment
The safety and efficacy of pegcetacoplan have not been studied in patients with hepatic impairment; however, no dose adjustment is recommended, as hepatic impairment is not expected to impact clearance of pegcetacoplan.
Paediatric population
The safety and efficacy of ASPAVELI in children with PNH aged 0 to <18 years have not yet been established. No data are available.
This medicinal product should not be used in children <12 years of age, as non-clinical safety data are not available for this age group.
Method of administration
ASPAVELI should only be administered via subcutaneous administration using a commercially available syringe system infusion pump. This medicinal product can be self-administered. When self-administration is initiated, the patient will be instructed by a qualified healthcare professional in infusion techniques, the use of a syringe system infusion pump, the keeping of a treatment record, the recognition of possible adverse reactions, and measures to be taken in case these occur.
ASPAVELI should be infused in the abdomen, thigh, or upper arms. Infusion sites should be at least 7.5 cm apart from each other. The infusion sites should be rotated between administration. Infusion into areas where the skin is tender, bruised, red, or hard should be avoided. Infusion into tattoos, scars, or stretch marks should be avoided. The typical infusion time is approximately 30 minutes (if using two sites) or approximately 60 minutes (if using one site). The infusion should be started promptly after drawing this medicinal product into the syringe. Administration should be completed within 2 hours after preparing the syringe. For instructions on the preparation and infusion of the medicinal product, see section 6.6.
Hypersensitivity to pegcetacoplan or to any of the excipients listed in section 6.1.
Pegcetacoplan therapy must not be initiated in patients:
• with unresolved infection caused by encapsulated bacteria including Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae (see section 4.4).
• who are not currently vaccinated against Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae unless they receive prophylactic treatment with appropriate antibiotics until 2 weeks after vaccination (see section 4.4).
Serious infections caused by encapsulated bacteria
The use of pegcetacoplan may predispose individuals to serious infections caused by encapsulated bacteria including Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae. To reduce the risk of infection, all patients must be vaccinated against these bacteria according to applicable local guidelines at least 2 weeks prior to receiving ASPAVELI, unless the risk of delaying therapy outweighs the risk of developing an infection.
Patients with known history of vaccination
Before receiving treatment with ASPAVELI, in patients with a known history of vaccination, it should be ensured that patients have received vaccines against encapsulated bacteria including Streptococcus pneumoniae, Neisseria meningitidis types A, C, W, Y, and B, and Haemophilus influenzae Type B within 2 years prior to starting ASPAVELI.
Patients without known history of vaccination
For patients without known history of vaccination, the required vaccines should be administered at least 2 weeks prior to receiving the first dose of ASPAVELI. If immediate therapy is indicated, the required vaccines should be administered as soon as possible and the patient treated with appropriate antibiotics until 2 weeks after vaccination.
Monitoring patients for serious infections
Vaccination may not be sufficient to prevent serious infection. Consideration should be given to official guidance on the appropriate use of antibacterial agents. All patients should be monitored for early signs of infections caused by encapsulated bacteria including Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae, evaluated immediately if infection is suspected, and treated with appropriate antibiotics if necessary. Patients should be informed of these signs and symptoms, and steps taken to seek medical care immediately. Physicians must discuss the benefits and risks of ASPAVELI therapy with patients.
Hypersensitivity
Hypersensitivity reactions have been reported. If a severe hypersensitivity reaction (including anaphylaxis) occurs, infusion with ASPAVELI must be discontinued immediately, and appropriate treatment instituted.
Injection site reactions
Injection site reactions have been reported with the use of subcutaneous ASPAVELI (see section 4.8). Patients should be trained appropriately in proper injection technique.
PNH laboratory monitoring
Patients with PNH receiving ASPAVELI should be monitored regularly for signs and symptoms of haemolysis, including measuring LDH levels, and may require dose adjustment within the recommended dosing schedule (see section 4.2).
Effects on laboratory tests
There may be interference between silica reagents in coagulation panels and pegcetacoplan that results in artificially prolonged activated partial thromboplastin time (aPTT); therefore, the use of silica reagents in coagulation panels should be avoided.
Treatment discontinuation for PNH
If patients with PNH discontinue treatment with ASPAVELI, they should be closely monitored for signs and symptoms of serious intravascular haemolysis. Serious intravascular haemolysis is identified by elevated LDH levels along with sudden decrease in PNH clone size or haemoglobin, or reappearance of symptoms such as fatigue, haemoglobinuria, abdominal pain, dyspnoea, major adverse vascular event (including thrombosis), dysphagia, or erectile dysfunction. If discontinuation of this medicinal product is necessary, alternate therapy should be considered. If serious haemolysis occurs after discontinuation, consider the following procedures/treatments: blood transfusion (packed RBCs), exchange transfusion, anticoagulation, and corticosteroids. Patients should be closely monitored for at least 8 weeks from the last dose, representing more than 5 half-lives of this medicinal product, to allow for medicinal product washout (see section 5.2) to detect serious haemolysis and other reactions. In addition, slow weaning should be considered.
Contraception in women of childbearing potential
It is recommended that women of childbearing potential use effective contraception methods to prevent pregnancy during treatment with pegcetacoplan and for at least 8 weeks after the last dose of pegcetacoplan (see section 4.6).
Polyethylene glycol (PEG) accumulation
ASPAVELI is a PEGylated medicinal product. The potential long term effects of PEG accumulation in the kidneys, the choroid plexus of the brain, and other organs are unknown (see section 5.3). Regular laboratory testing of renal function is recommended.
Educational materials
All physicians who intend to prescribe ASPAVELI must ensure they have received and are familiar with the physician educational material. Physicians must explain and discuss the benefits and risks of ASPAVELI therapy with the patient and provide them with the patient information pack and the patient card. The patient should be instructed to seek prompt medical care if they experience any sign or symptom of serious infection or hypersensitivity during therapy with ASPAVELI, especially if indicative of infection with encapsulated bacteria.
Excipients with known effect
Sorbitol content
ASPAVELI 1 080 mg contains 820 mg sorbitol in each vial.
Patients with hereditary fructose intolerance (HFI) should not take/be given this medicinal product.
Sodium content
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, that is to say, essentially 'sodium-free'.
No interaction studies have been performed. Based on in vitro data, pegcetacoplan has low potential for clinical drug-drug interactions.
Women of childbearing potential
It is recommended that women of childbearing potential use effective contraception methods to prevent pregnancy during treatment with pegcetacoplan and for at least 8 weeks after the last dose of pegcetacoplan. For women planning to become pregnant, the use of ASPAVELI may be considered following an assessment of the risks and benefits (see Pregnancy).
Pregnancy
There are no or limited amount of data from the use of pegcetacoplan in pregnant women. Studies in animals have shown reproductive toxicity (see section 5.3).
ASPAVELI is not recommended during pregnancy and in women of childbearing potential not using contraception.
Breast-feeding
It is unknown whether pegcetacoplan is excreted in human milk. The potential for absorption and harm to the breastfed infant is unknown. Animal data suggest a low excretion (less than 1%, not pharmacologically significant) of pegcetacoplan in monkey milk (see section 5.3). It is unlikely that a breastfed infant would have clinically relevant exposure.
It is recommended to discontinue breast-feeding during pegcetacoplan treatment.
Fertility
No animal or human data on the effect of pegcetacoplan on fertility are available. In toxicity studies, there were no microscopic abnormalities in male or female reproductive organs in monkeys (see section 5.3).
ASPAVELI has no or negligible influence on the ability to drive and use machines.
Summary of the safety profile
The most commonly reported adverse reactions in patients treated with ASPAVELI were injection site reactions: injection site erythema, injection site pruritus, injection site swelling, injection site pain, injection site bruising. Other adverse reactions reported in more than 10% of patients during clinical studies were upper respiratory tract infection, diarrhoea, haemolysis, abdominal pain, headache, fatigue, and pyrexia, cough, urinary tract infection, vaccination complication, dizziness, pain in extremity, arthralgia, back pain, nausea. The most commonly reported serious adverse reactions were haemolysis and sepsis.
Tabulated list of adverse reactions
Table 1 gives the adverse reactions observed from the clinical studies with pegcetacoplan in patients with PNH. Adverse reactions are listed by MedDRA system organ class (SOC) and frequency, using the following convention: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1 000 to <1/100) or rare (≥1/10 000 to <1/1 000), very rare (<1/10 000), and not known (cannot be estimated from available data).
Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
Table 1: Adverse reactions
|
MedDRA System Organ Class |
Frequency |
Adverse reaction |
|
Infections and infestations |
Very common |
Upper respiratory tract infection Urinary tract infection |
|
Common |
Sepsis Gastrointestinal infection Fungal infection Skin infection Oral infection Ear infection Infection Respiratory tract infection Viral infection Bacterial infection Hordeolum | |
|
Uncommon |
COVID-19 Cervicitis Groin infection Pneumonia Nasal abscess Ophthalmic herpes zoster Vulvovaginal mycotic infection | |
|
Blood and lymphatic system disorders |
Very common |
Haemolysis |
|
Common |
Thrombocytopenia Neutropenia | |
|
Metabolism and nutrition disorders |
Common |
Hypokalaemia |
|
Psychiatric disorders |
Common |
Anxiety |
|
Nervous system disorders |
Very common |
Headache Dizziness |
|
Vascular disorders |
Common |
Hypertension |
|
Respiratory, thoracic and mediastinal disorders |
Very common |
Cough |
|
Common |
Dyspnoea Epistaxis Oropharyngeal pain Nasal congestion | |
|
Gastrointestinal disorders |
Very common |
Abdominal pain Diarrhoea Nausea |
|
Skin and subcutaneous tissue disorders |
Common |
Erythema Rash |
|
Musculoskeletal and connective tissue disorders |
Very common |
Arthralgia Back pain Pain in extremity |
|
Common |
Myalgia Muscle spasms | |
|
Renal and urinary disorders |
Common |
Acute kidney injury Chromaturia |
|
General disorders and administration site conditions |
Very common |
Injection site erythema Injection site pruritus Injection site swelling Injection site bruising Fatigue Pyrexia Injection site pain |
|
Common |
Injection site reaction Injection site induration | |
|
Investigations |
Common |
Alanine aminotransferase increased Bilirubin increased |
|
Injury, poisoning and procedural complications |
Very common |
Vaccination complication1 |
The adverse reactions listed in the table are from clinical studies APL2-302, Study 202, Study 204, and Study CP0514 in PNH.
Medically similar terms are grouped, where appropriate, on the basis of similar medical concept.
1Vaccination complications were related to the mandatory vaccinations.
Description of selected adverse reactions
Infections
Based on its mechanism of action, the use of pegcetacoplan may potentially increase the risk of infections, particularly infections caused by encapsulated bacteria including Streptococcus pneumoniae, Neisseria meningitidis types A, C, W, Y, and B, and Haemophilus influenzae (see section 4.4). No serious infection caused by encapsulated bacteria was reported during Study APL2-302. Forty-eight patients experienced an infection during the study. The most frequent infections in patients treated with pegcetacoplan during Study APL2-302 were upper respiratory tract infection (28 cases, 35%). Most infections reported in patients treated with pegcetacoplan during study APL2-302 were non-serious, and predominantly mild in intensity. Ten patients developed infections reported as serious including one patient who died due to COVID-19. The most frequent serious infections were sepsis (3 cases) (leading to discontinuation of pegcetacoplan in one patient) and gastroenteritis (3 cases); all of which resolved.
Haemolysis
Nineteen patients reported haemolysis during Study APL2-302 in patients treated with pegcetacoplan. Seven cases were reported as serious, and 5 cases led to discontinuation of pegcetacoplan and the dose of pegcetacoplan was increased in 10 patients.
Immunogenicity
Anti-drug antibody (ADA) incidence (seroconverted ADA or boosted ADA from pre-existing level) were low, and when present, had no noticeable impact on the PK/PD, efficacy, or safety profile of pegcetacoplan. Throughout Study APL2-302, 2 out of 80 patients developed anti-pegcetacoplan peptide antibodies. Both patients also tested positive for neutralizing antibody (NAb). NAb response had no apparent impact on PK or clinical efficacy. Six out of 80 patients developed anti-PEG antibody incidence; 2 were seroconversions and 4 were treatment-boosted.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme.
Website: www.mhra.gov.uk/yellowcard or search for MHRA Yellow Card in the Google Play or Apple App Store.
No case of overdose has been reported to date. In case of overdose, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions and appropriate symptomatic treatment be instituted.
Pharmacotherapeutic group: Immunosuppressants, Selective immunosuppressants, ATC code: L04AA54
Mechanism of action
Pegcetacoplan is a symmetrical molecule comprised of two identical pentadecapeptides covalently bound to the ends of a linear 40-kDa PEG molecule. The peptide moieties bind to complement C3 and exert a broad inhibition of the complement cascade. The 40-kDa PEG moiety imparts improved solubility and longer residence time in the body after administration of the medicinal product.
Pegcetacoplan binds to complement protein C3 and its activation fragment C3b with high affinity, thereby regulating the cleavage of C3 and the generation of downstream effectors of complement activation. In PNH, extravascular haemolysis (EVH) is facilitated by C3b opsonization while intravascular haemolysis (IVH) is mediated by the downstream membrane attack complex (MAC). Pegcetacoplan exerts broad regulation of the complement cascade by acting proximal to both C3b and MAC formation, thereby controlling the mechanisms that lead to EVH and IVH.
Pharmacodynamic effects
In Study APL2-302, mean C3 concentration increased from 0.94 g/L at baseline to 3.83 g/L at Week 16 in the pegcetacoplan group. The baseline percentage of PNH Type II + III RBCs was 66.80%, which then increased to 93.85% at Week 16. The mean percentage of PNH Type II + III RBCs with C3 deposition was 17.73% at baseline and this decreased to 0.20% at Week 16.
Clinical efficacy and safety
The efficacy and safety of ASPAVELI in patients with PNH was assessed in a phase 3 study (APL2-302) in which an open-label, randomised, active comparator-controlled period of 16 weeks was followed by a 32-week open label period (OLP). This study enrolled patients with PNH who had been treated with a stable dose of eculizumab for at least the previous 3 months and with haemoglobin levels <10.5 g/dL.
Study APL2-302
The dose of ASPAVELI was 1 080 mg twice weekly. Eligible patients entered a 4-week run-in period during which they received ASPAVELI 1 080 mg subcutaneously twice weekly in addition to their current dose of eculizumab. Patients were then randomised in a 1:1 ratio to receive either 1 080 mg of ASPAVELI twice weekly or their current dose of eculizumab through the duration of the 16-week randomised controlled period (RCP). Randomisation was stratified based on the number of packed red blood cell (PRBC) transfusions within the 12 months prior to Day -28 (<4; ≥4) and platelet count at screening (<100 000/mm3; ≥100 000/mm3). Patients who completed the RCP entered the OLP during which all patients received ASPAVELI for up to 32 weeks (patients who received eculizumab during the RCP entered a 4-week run-in period before switching to ASPAVELI monotherapy). If required, the dose of ASPAVELI could be adjusted to 1 080 mg every 3 days.
The primary and secondary efficacy endpoints were assessed at Week 16. The primary efficacy endpoint was change from Baseline to Week 16 (during RCP) in haemoglobin level. Baseline was defined as the average of measurements prior to the first dose of pegcetacoplan (at the beginning of the run-in period). Key secondary efficacy endpoints were transfusion avoidance, defined as the proportion of patients who did not require a transfusion during the RCP, and change from Baseline to Week 16 in absolute reticulocyte count (ARC), LDH level, and FACIT-Fatigue scale score.
A total of 80 patients entered the run-in period. At the end of the run-in period, all 80 were randomised, 41 to ASPAVELI and 39 to eculizumab. Demographics and baseline disease characteristics were generally well balanced between treatment groups (see Table 2). A total of 38 patients in the group treated with ASPAVELI and 39 patients in the eculizumab group completed the 16-week RCP and continued into the 32-week open-label period. In total, 12 of 80 (15%) patients receiving ASPAVELI discontinued due to adverse events. Per protocol 15 patients had their dose adjusted to 1 080 mg every 3 days. Twelve patients were evaluated for benefit and 8 of the 12 patients demonstrated benefit from the dose adjustment.
Table 2: Patient baseline demographics and characteristics in Study APL2-302
|
Parameter |
Statistics |
ASPAVELI (N=41) |
Eculizumab (N=39) |
|
Age (years) 18-64 years ≥65 years |
Mean (SD) n (%) n (%) |
50.2 (16.3) 31 (75.6) 10 (24.4) |
47.3 (15.8) 32 (82.1) 7 (17.9) |
|
Dose level of eculizumab at baseline Every 2 weeks IV 900 mg Every 11 days IV 900 mg Every 2 weeks IV 1 200 mg Every 2 weeks IV 1 500 mg |
n (%) n (%) n (%) n (%) |
26 (63.4) 1 (2.4) 12 (29.3) 2 (4.9) |
29 (74.4) 1 (2.6) 9 (23.1) 0 |
|
Female |
n (%) |
27 (65.9) |
22 (56.4) |
|
Time since diagnosis of PNH (years) to Day -28 |
Mean (SD) |
8.7 (7.4) |
11.4 (9.7) |
|
Haemoglobin level (g/dL) |
Mean (SD) |
8.7 (1.1) |
8.7 (0.9) |
|
Reticulocyte count (109/L) |
Mean (SD) |
218 (75.0) |
216 (69.1) |
|
LDH level (U/L) |
Mean (SD) |
257.5 (97.6) |
308.6 (284.8) |
|
Total FACIT-Fatigue* |
Mean (SD) |
32.2 (11.4) |
31.6 (12.5) |
|
Number of transfusions in last 12 months prior to Day -28 |
Mean (SD) |
6.1 (7.3) |
6.9 (7.7) |
|
<4 |
n (%) |
20 (48.8) |
16 (41.0) |
|
≥4 |
n (%) |
21 (51.2) |
23 (59.0) |
|
Platelet count at screening (count/mm3) |
Mean (SD) |
167 (98.3) |
147 (68.8) |
|
<100 000 |
n (%) |
12 (29.3) |
9 (23.1) |
|
≥100 000 |
n (%) |
29 (70.7) |
30 (76.9) |
|
History of aplastic anaemia |
n (%) |
11 (26.8) |
9 (23.1) |
|
History of myelodysplastic syndrome |
n (%) |
1 (2.4) |
2 (5.1) |
*FACIT-Fatigue is measured on a scale of 0-52, with higher values indicating less fatigue.
ASPAVELI was superior to eculizumab for the primary endpoint of the haemoglobin change from baseline (P<0.0001).
Figure 1. Adjusted mean change in haemoglobin (g/dL) from baseline to Week 16
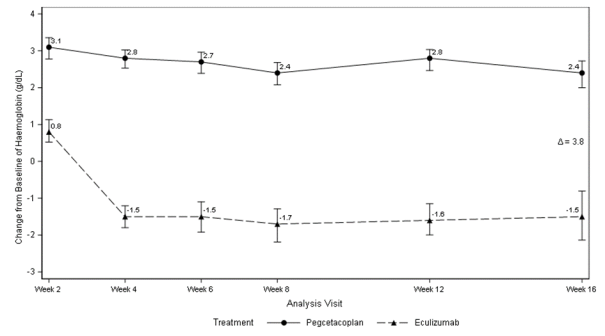
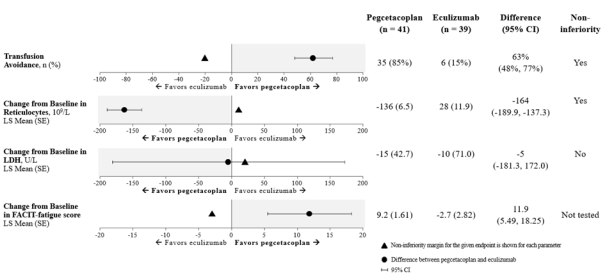
Non-inferiority was demonstrated in key secondary endpoints of transfusion avoidance and change from baseline in ARC.
Non-inferiority was not met in change from baseline in LDH.
Due to hierarchical testing, statistical testing for change from baseline for FACIT-Fatigue score was not formally tested.
The adjusted means, treatment difference, confidence intervals, and statistical analyses performed for the key secondary endpoints are shown in Figure 2.
Figure 2. Key secondary endpoints analysis
Results were consistent across all supportive analyses of the primary and key secondary endpoints, including all observed data with post transfusion data included.
Haemoglobin normalization was achieved in 34% of patients in the ASPAVELI group versus 0% in the eculizumab group at Week 16. LDH normalization was achieved in 71% of patients in the group treated with ASPAVELI versus 15% in the eculizumab group.
A total of 77 patients entered the 32-week OLP, during which all patients received ASPAVELI, resulting in a total exposure of up to 48 weeks. The results at Week 48 were generally consistent with those at Week 16 and support sustained efficacy.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with ASPAVELI in one or more subsets of the paediatric population in paroxysmal nocturnal haemoglobinuria (see section 4.2 for information on paediatric use).
Absorption
Pegcetacoplan is administered by subcutaneous infusion and gradually absorbed into the systemic circulation with a median Tmax between 108 and 144 hours (4.5 to 6.0 days) following a single subcutaneous dose to healthy volunteers. Steady-state serum concentrations following twice weekly dosing at 1 080 mg in patients with PNH were achieved approximately 4 to 6 weeks following the first dose and mean (%CV) steady-state serum concentrations ranged between 655 (18.6%) to 706 (15.1%) µg/mL in patients treated for 16 weeks. Steady state concentrations in the patients (n=22) that continued to receive pegcetacoplan up to Week 48 were 622.94 µg/mL (39.7%), indicating sustainable therapeutic concentrations of pegcetacoplan through Week 48. The bioavailability of a subcutaneous dose of pegcetacoplan is estimated to be 77% based on population PK analysis.
Distribution
The mean (%CV) volume of distribution of pegcetacoplan is approximately 3.9 L (35%) in patients with PNH based on population PK analysis.
Metabolism/elimination
Based on its PEGylated peptide structure, the metabolism of pegcetacoplan is expected to occur via catabolic pathways and be degraded into small peptides, amino acids, and PEG. Results of a radiolabelled study in cynomolgus monkeys suggest the primary route of elimination of the labelled peptide moiety is via urinary excretion. Although the elimination of PEG was not studied, it is known to undergo renal excretion.
Pegcetacoplan showed no inhibition or induction of the CYP enzyme isoforms tested as demonstrated from the results of in vitro studies. Pegcetacoplan was neither a substrate nor an inhibitor of the human uptake or efflux transporters.
Following multiple subcutaneous dosing of pegcetacoplan in patients with PNH, the mean (%CV) of clearance is 0.015 (28%) L/h and median effective half-life of elimination (t1/2) is 8.0 days as estimated by the population PK analysis.
Linearity/non-linearity
Exposure of pegcetacoplan increases in a dose proportional manner from 45 to 1 440 mg.
Special populations
No impact on the pharmacokinetics of pegcetacoplan was identified with age (19-81 years) and sex based on the results of population PK analysis. Race was also shown not to have an impact; however, data are limited and therefore not considered conclusive.
Patients with a body weight below 50 kg are predicted to have up to 34% higher average exposure at steady state compared to a 70-kg subject, based on population PK analysis. Minimal data are available on the safety profile of pegcetacoplan for patients with a body weight below 50 kg.
Elderly
Although there were no apparent age-related differences observed in these studies, the number of patients aged 65 years and over is not sufficient to determine whether they respond differently from younger patients. See section 4.2.
Renal impairment
In a study of 8 patients with severe renal impairment, defined as creatinine clearance (CrCl) less than 30 mL/min using the Cockcroft-Gault formula (with 4 patients with values less than 20 mL/min), renal impairment had no effect on the pharmacokinetics of a single 270-mg dose of pegcetacoplan. There are minimal data on patients with PNH with renal impairment who have been administered the clinical dose of 1 080 mg twice weekly. There are no available clinical data for the use of pegcetacoplan in patients with ESRD requiring haemodialysis. See section 4.2.
In vitro and in vivo toxicology data reveal no toxicity of special concern for humans. Effects observed in animals at exposure levels similar to clinical exposure levels are described below. These effects were not observed in clinical studies.
Animal reproduction
Pegcetacoplan treatment of pregnant cynomolgus monkeys at a subcutaneous dose of 28 mg/kg/day (2.9 times the human steady-state Cmax) from the gestation period through parturition resulted in a statistically significant increase in abortions or stillbirths. No maternal toxicity or teratogenic effects were observed in offspring delivered at term. Additionally, no developmental effects were observed in infants up to 6 months postpartum. Systemic exposure to pegcetacoplan was detected in foetuses from monkeys treated with 28 mg/kg/day from the period of organogenesis through the second trimester, but the exposure was minimal (less than 1%, not pharmacologically significant).
Carcinogenesis
Long term animal carcinogenicity studies of pegcetacoplan have not been conducted.
Genotoxicity
Pegcetacoplan was not mutagenic when tested in in vitro bacterial reverse mutation (Ames) assays and was not genotoxic in an in vitro assay in human TK6 cells or in an in vivo micronucleus assay in mice.
Animal toxicology
Repeat-dose studies were conducted in rabbits and cynomolgus monkeys with daily subcutaneous doses of pegcetacoplan up to 7 times the human dose (1 080 mg twice weekly). Histologic findings in both species included dose-dependent epithelial vacuolation and infiltrates of vacuolated macrophages in multiple tissues. These findings have been associated with large cumulative doses of long-chain PEG in other marketed PEGylated drugs, were without clinical consequence, and were not considered adverse. Reversibility was not demonstrated in the pegcetacoplan animal studies after one month and was not evaluated for a longer duration. Data from literature suggest reversibility of PEG vacuoles.
Renal tubular degeneration was observed microscopically in both species at exposures (Cmax and AUC) less than or comparable to those for the human dose and was minimal and nonprogressive between 4 weeks and 9 months of daily administration of pegcetacoplan. Although no overt signs of renal dysfunction were observed in animals, the clinical significance and functional consequence of these findings are unknown.
Sorbitol (E 420)
Glacial acetic acid
Sodium acetate trihydrate
Sodium hydroxide (for pH adjustment)
Water for injection
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
2 years.
Store in a refrigerator (2 °C – 8 °C).
Store in the original carton to protect from light.
A Type I glass vial with a stopper (cholorobutyl), and a seal (aluminium) with a flip-off cap (polypropylene) containing 54 mg/mL of sterile solution.
Each single pack contains 1 vial.
Multipack containing 8 (8 packs of 1) vials.
Not all pack sizes may be marketed.
ASPAVELI comes as a ready-to-use solution in single-use vials. Because the solution contains no preservative, this medicinal product should be infused immediately after preparing the syringe.
ASPAVELI is a clear, colourless to slightly yellowish aqueous solution. Do not use if the liquid looks cloudy, contains particles, or is dark yellow.
Always bring the vial to the room temperature for approximately 30 minutes before use.
Remove the protective flip cap from the vial to expose the central portion of the gray rubber stopper of the vial. Clean the stopper with a new alcohol wipe and allow the stopper to dry. Do not use if the protective flip cap is missing or damaged.
Option 1: If using a needleless transfer device (such as a vial adapter), follow the instructions provided by the device manufacturer.
Option 2: If transfer is done using a transfer needle and a syringe, follow the instructions below:
• Attach a sterile transfer needle to a sterile syringe.
• Pull back the plunger to fill the syringe with air, which should be about 20 mL.
• Make sure the vial is in upright position. Do not turn the vial upside down.
• Push the air-filled syringe with transfer needle attached through the centre of the vial stopper.
• The tip of the transfer needle should not be in the solution to avoid creating bubbles.
• Gently push the air from the syringe into the vial. This will inject the air from the syringe into the vial.
• Invert the vial.
• With the transfer needle tip in the solution, slowly pull the plunger to fill the syringe with all the liquid.
• Remove the filled syringe and the transfer needle from the vial.
• Do not recap the transfer needle. Unscrew the needle and throw it away in the sharps container.
Follow the device manufacturer's instructions to prepare the infusion pump and tubing.
Potential areas for infusion include the abdomen, thighs, hips, or upper arms. Rotate infusion sites from one infusion to the next. If there are multiple infusion sites, they should be at least 7.5 cm apart.
The typical infusion time is approximately 30 minutes (if using two sites) or approximately 60 minutes (if using one site).
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
Swedish Orphan Biovitrum AB (publ)
SE 112 76 Stockholm
Sweden
PLGB 30941/0022
24/02/2022
14/09/2022
